问题描述
我已经共享了一个 Rscript 来在 linux 集群上运行,这是我以前从未做过的事情(现在由于 covid 限制,没有人可以调试它)。该脚本从 R getopt 包开始,其中似乎是矩阵下方的 if 选项:
library("getopt")
optspec <- matrix(c(
'reads','r',1,'character','/path/to/fastq/reads/','out_dir','o','Path to output directory','aggregate','a','logical','Produce aggregated plots','help','h','Display help'
),byrow=TRUE,ncol=5)
opt <- getopt(optspec)
if (!is.null(opt$help)) {
cat(getopt(optspec,usage=TRUE))
q(status=1)
}
if (is.null(opt$reads)) {
cat("Error: no reads argument provconda ided\n")
cat(getopt(optspec,usage=TRUE))
q(status=1)
}
if (is.null(opt$out_dir)) {
cat("Error: no out_dir argument provided\n")
cat(getopt(optspec,usage=TRUE))
q(status=1)
}
aggregate=FALSE
if (!is.null(opt$aggregate)) {
aggregate=TRUE
}
我在集群生成的输出文件中返回的错误总是:
错误:没有读取参数provconda ided
这就是为什么我一直专注于代码行
'reads',我尝试过以各种方式编辑 fastq 文件的路径(脚本最初表示“读取”、“r”、“1”、“字符”、“输入目录路径”)我已经看到 R 和linux(引号,开头没有 /,用 . 替换路径等)但仍然出现相同的错误,所以我现在不知道是什么导致了它。我确定它很容易我遗漏/不理解,但我无法发现我哪里出错了。
脚本以
开头#!/usr/bin/env Rscript
#$ -j y
#$ -cwd
并且是从我的 cwd 中 qsubbed 的,这是我需要分析的 fastq 文件所在的位置。我在集群上的dada2环境中运行,因为这是序列分析协议的一部分。 感谢您提供的任何帮助。
解决方法
上面的代码不是脚本需要的选项设置的地方。参数需要从命令行传入。
您需要使用 my_script_name 或 my_script_name -r /path/to/fastq/reads 而不是使用 my_scrip_name --reads /path/to/fastq/reads 运行脚本
您显示的代码是脚本了解要接受哪些选项以及必须设置哪些选项的地方。不要编辑这些。
以
为例'reads','r',1,'character','/path/to/fastq/reads/'
前两个条目是选项名称的长短形式(reads 或 r),'r' 后面的 1 表示该选项后面需要一个字符类型的参数,每行的最后一个条目是帮助信息。
上面代码中的if语句的意思是如果你不给-r(或--reads)和-o(或--out_dir),脚本会打印帮助信息(你也可以通过调用带有 -h 作为选项的脚本)。 q(status=1) 是导致脚本不做任何事情就返回的原因。
-a(或 --aggragate)是可选的,默认为 FALSE 。如果您通过在命令行末尾添加“-a”来为脚本提供此选项,那么它将以某种方式更改您显示的代码未显示的绘图样式。
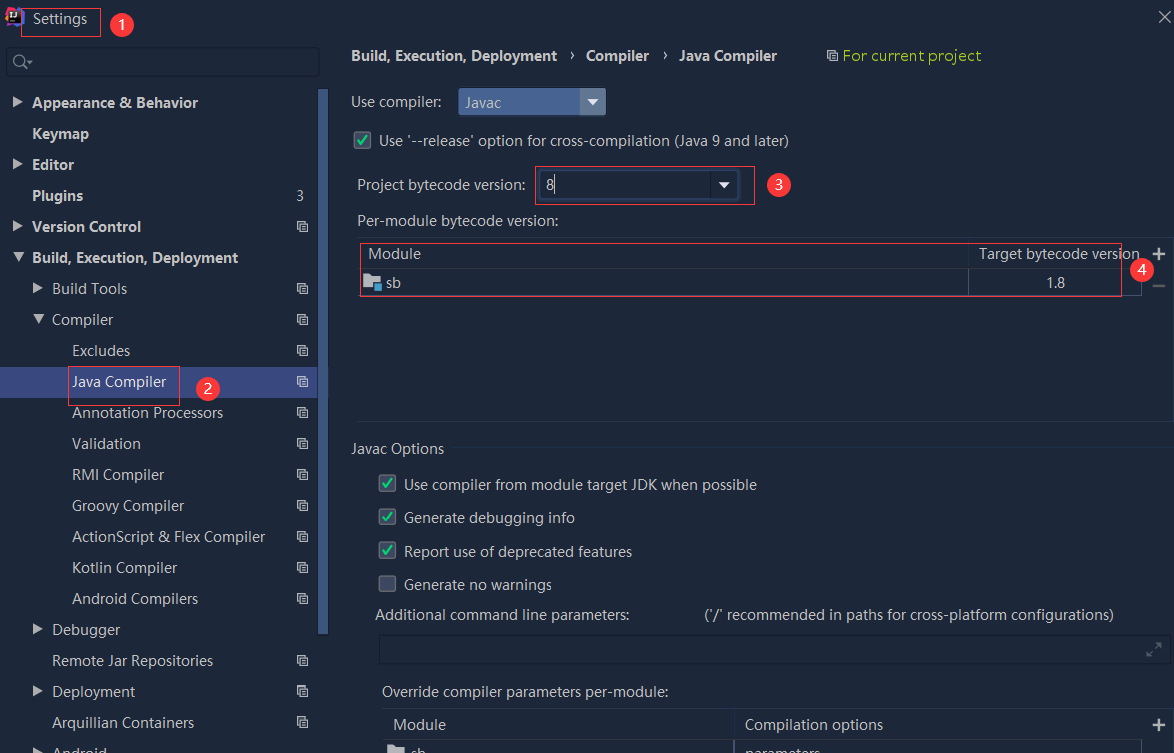 依赖报错 idea导入项目后依赖报错,解决方案:https://blog....
依赖报错 idea导入项目后依赖报错,解决方案:https://blog....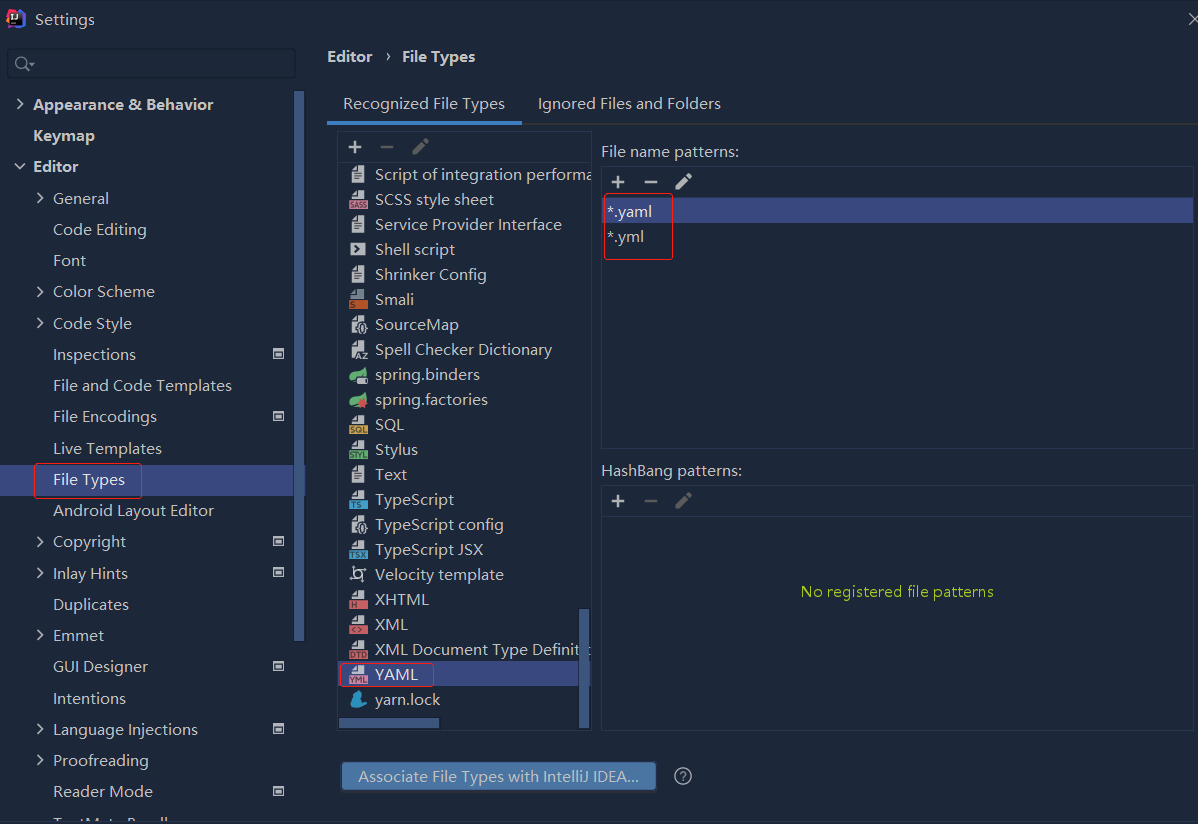
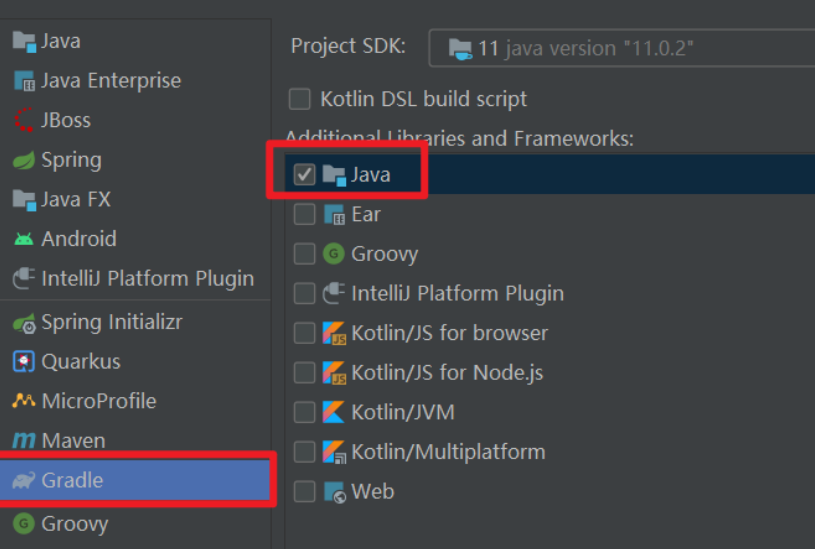 错误1:gradle项目控制台输出为乱码 # 解决方案:https://bl...
错误1:gradle项目控制台输出为乱码 # 解决方案:https://bl...